* What can do Cov-HiPathia for you
* How to use Cov-HiPathia:
* Tools:
* Worked examples:
* For further learning:
* What can do Cov-HiPathia for you
* How to use Cov-HiPathia:
* Tools:
* Worked examples:
* For further learning:
The Variant interpreter allows us to infer how a gene (or combination of genes) knockdown may deregulate the signaling pathways activity. This impact is evaluated by comparing the normal activity values from a given tissue, with the activity values of that same tissue calculated after the in silico knockdown of the expression values of the genes of interest.
This can be done for each signaling pathwat and for each one of the tissues included in GTEx. Additionally, the user can upload a file with gene expression data to perform the analysis on custom tissue.
Essentially, we are comparing pathway activities between two groups (Normal tissue Vs Knockdown tissue), to aid us in the functional interpretetion of disruptive genomic variants.
The tool can be accessed from the upper main menu bar, by clicking on the Variant interpreter button. See Workflow for further information.
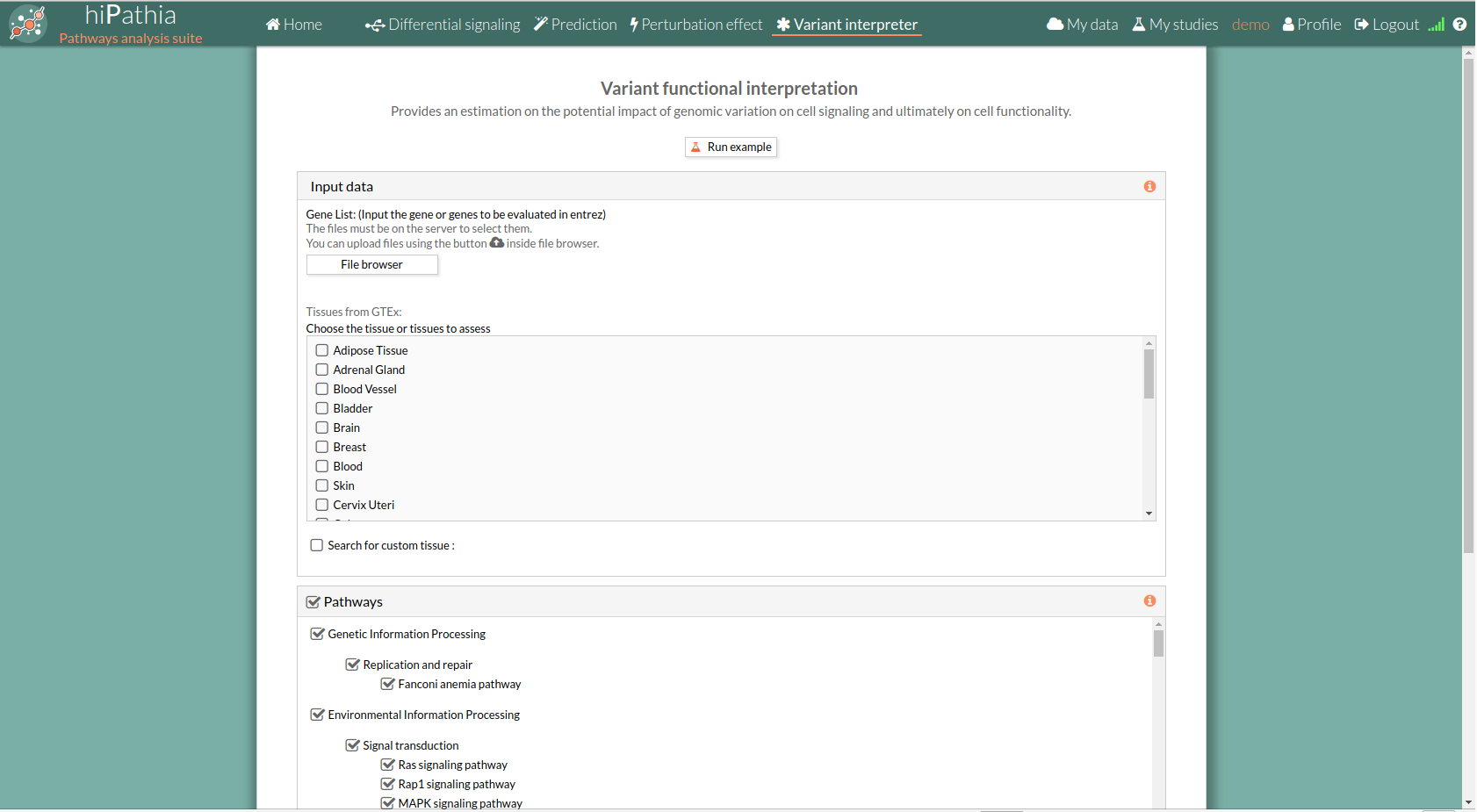
The main page of the tool is its filling in form. This form includes all the information and parameters that the tool needs to carry out the analysis. The form is divided in different panels:
 A tabulated text file with data on genomic variation must be uploaded. This file must include list of gene names in HUGO or entrez id (see gene list file format for more information).This genes list corresponds to the genes to in silico knockdown simultaneously.
A tabulated text file with data on genomic variation must be uploaded. This file must include list of gene names in HUGO or entrez id (see gene list file format for more information).This genes list corresponds to the genes to in silico knockdown simultaneously.
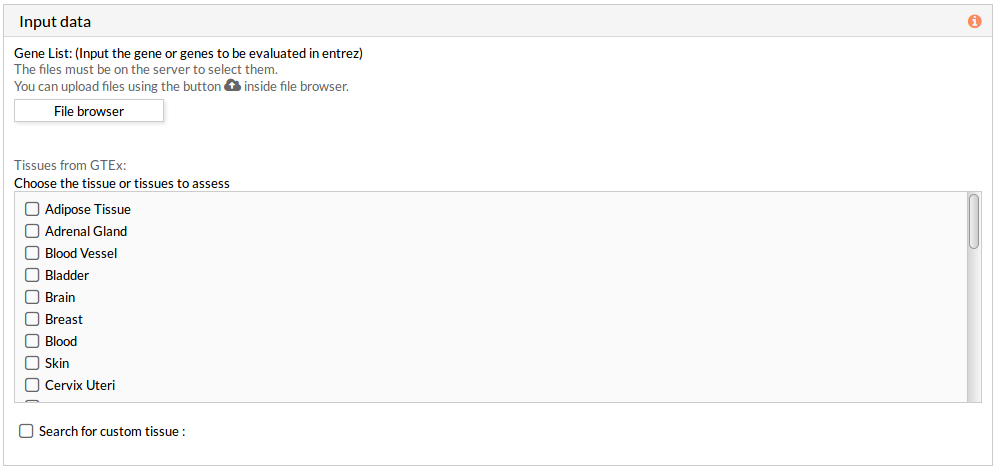
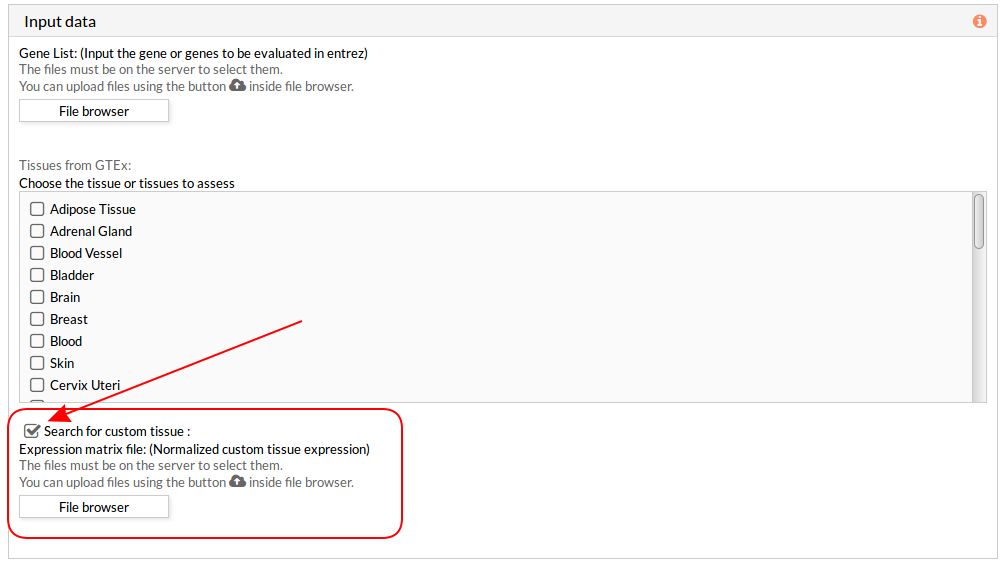
Then you have to select at least one tissue, or a custom matrix file has to be loaded after checking the “Search for custom tissue” checkbox as shown below:
 Note: You can select all options, but you have to take into account that the analysis will take more minutes depending on you custom tissue file and the server availability.
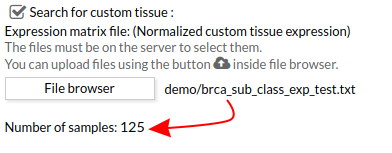
When you select the custom gene expression file, the number of samples of this matrix will appear under the “File browser” button as shown below.
Note: You can select all options, but you have to take into account that the analysis will take more minutes depending on you custom tissue file and the server availability.
When you select the custom gene expression file, the number of samples of this matrix will appear under the “File browser” button as shown below.
This panel includes the list of all available pathways in HiPathia. We can select the pathways with which the analysis will be performed.
HiPathia retrieves pathway information from KEGG database. KEGG pathway database is a collection of manually drawn pathway maps representing the knowledge on the molecular interaction, reaction and relation networks.
 By default all available pathways are selected.
By default all available pathways are selected.
Note: At least one pathway has to be selected.
This panel includes some parameters in order to identify and save our study.
Once the form has been filled in, press the Run analysis button to launch the study.
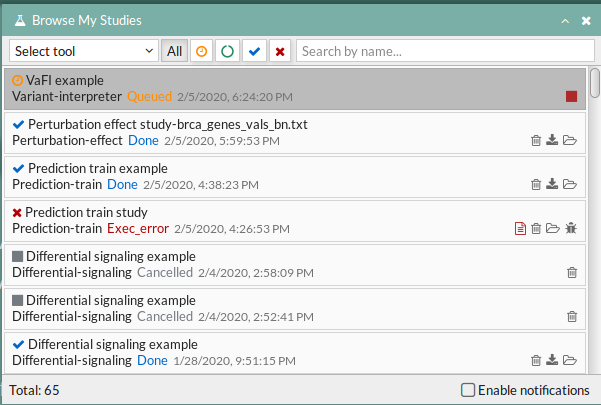
 Your study will be listed in the My studies panel, and a panel called Browse my studies will appear showing all your studies and their state. the new study will appear with a queued state then running state. If everything goes well, the state will be done after few minutes(depending on the inputs data and the availability of server).
All study states are:
Your study will be listed in the My studies panel, and a panel called Browse my studies will appear showing all your studies and their state. the new study will appear with a queued state then running state. If everything goes well, the state will be done after few minutes(depending on the inputs data and the availability of server).
All study states are:

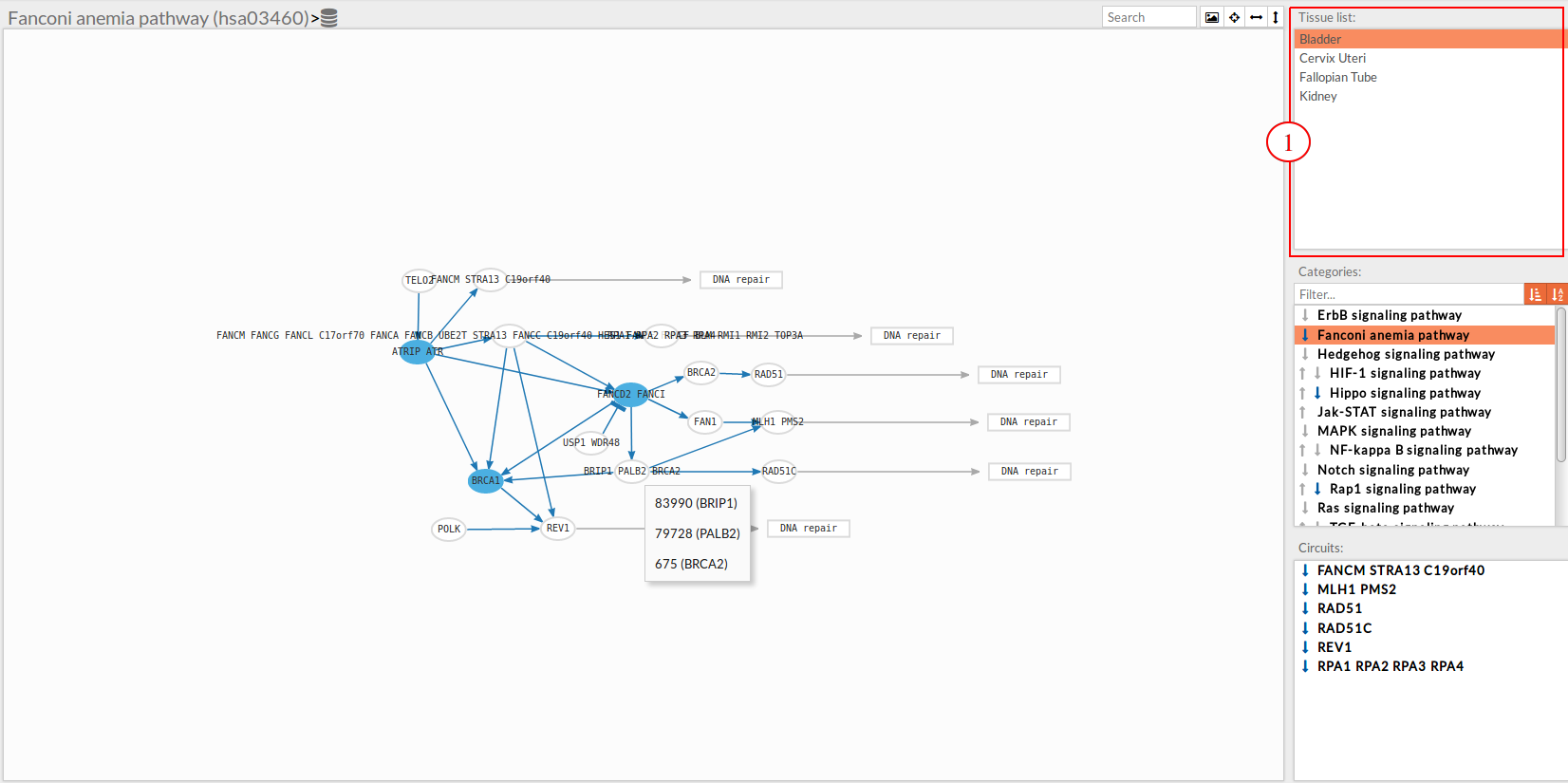
The results page of the Variant Functional Interpreter tool is an interactive working environment. You can simulate knockdown of one or several genes in the activity of signaling pathways. Essentially, it is comparing pathway activities between two groups (Normal tissue Vs Knockdown tissue), also you can study the effect of a Knockdown for several tissues simultaneously and compare between them. This interactive working environment is divided in different panels:
A list with selected tissues from GTEx. If the user uploaded a file with gene expression data a custom tissue will appear in this list under the name “Custom”.
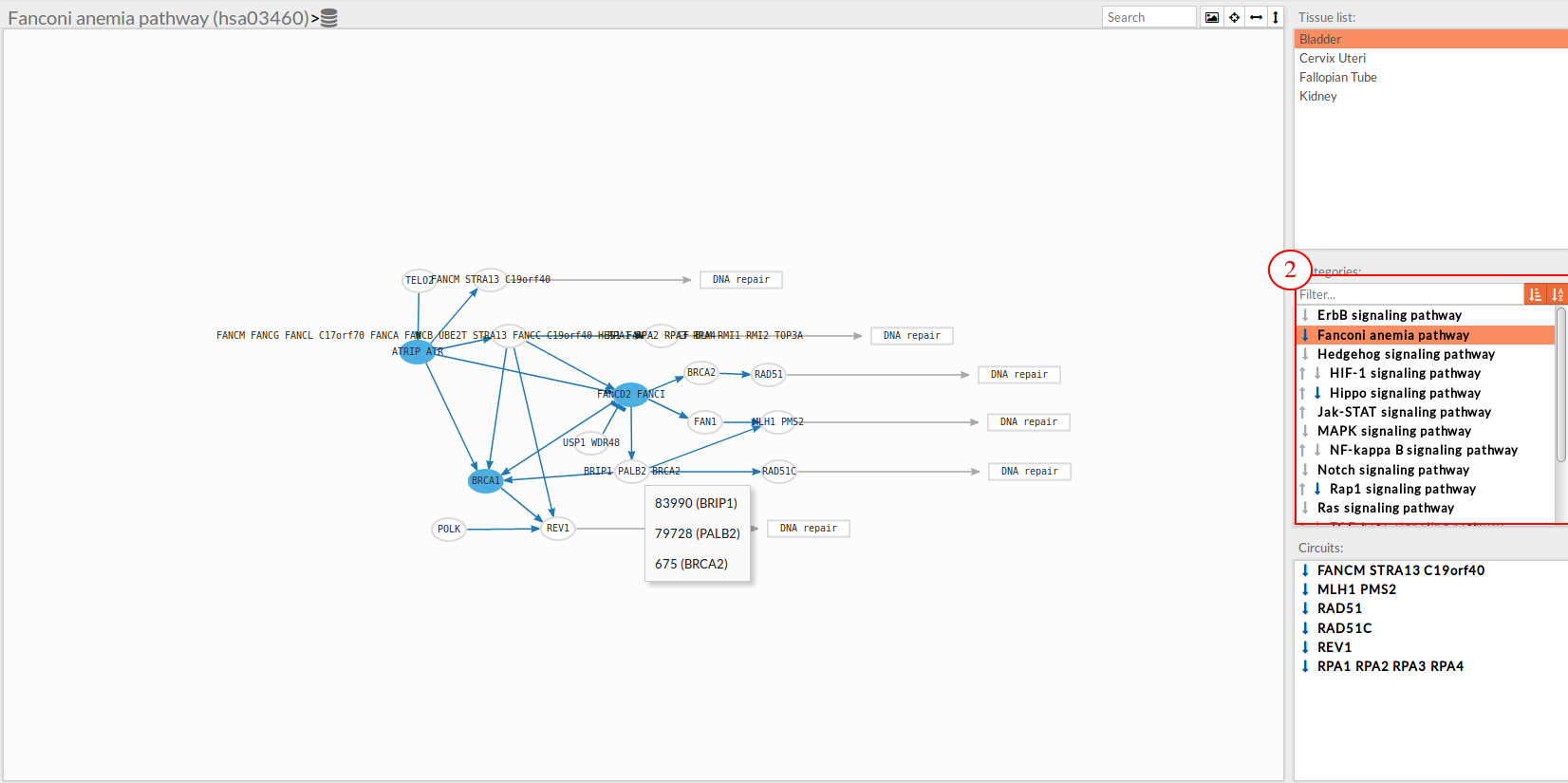
In this part of the visualization tool, all selected pathways from the variant interpreter form are shown along with one or two arrows. These arrows indicate whether in one of the “Effector circuit” within each pathway a differential activation pattern between the two compared groups (Normal tissue Vs Knockdown tissue).
The arrows will be colored if the differential activation is significant after the p-value adjustment (calculated from the circuit activity values using Wilcoxon test for paired data) and depicted in grey if it isn't.
The arrow will point up or down if an up-regulation or down-regulation of the signal occurs between the circuits within that pathway.
Only one arrow is shown if all the effector circuits are whether up or down-regulated.
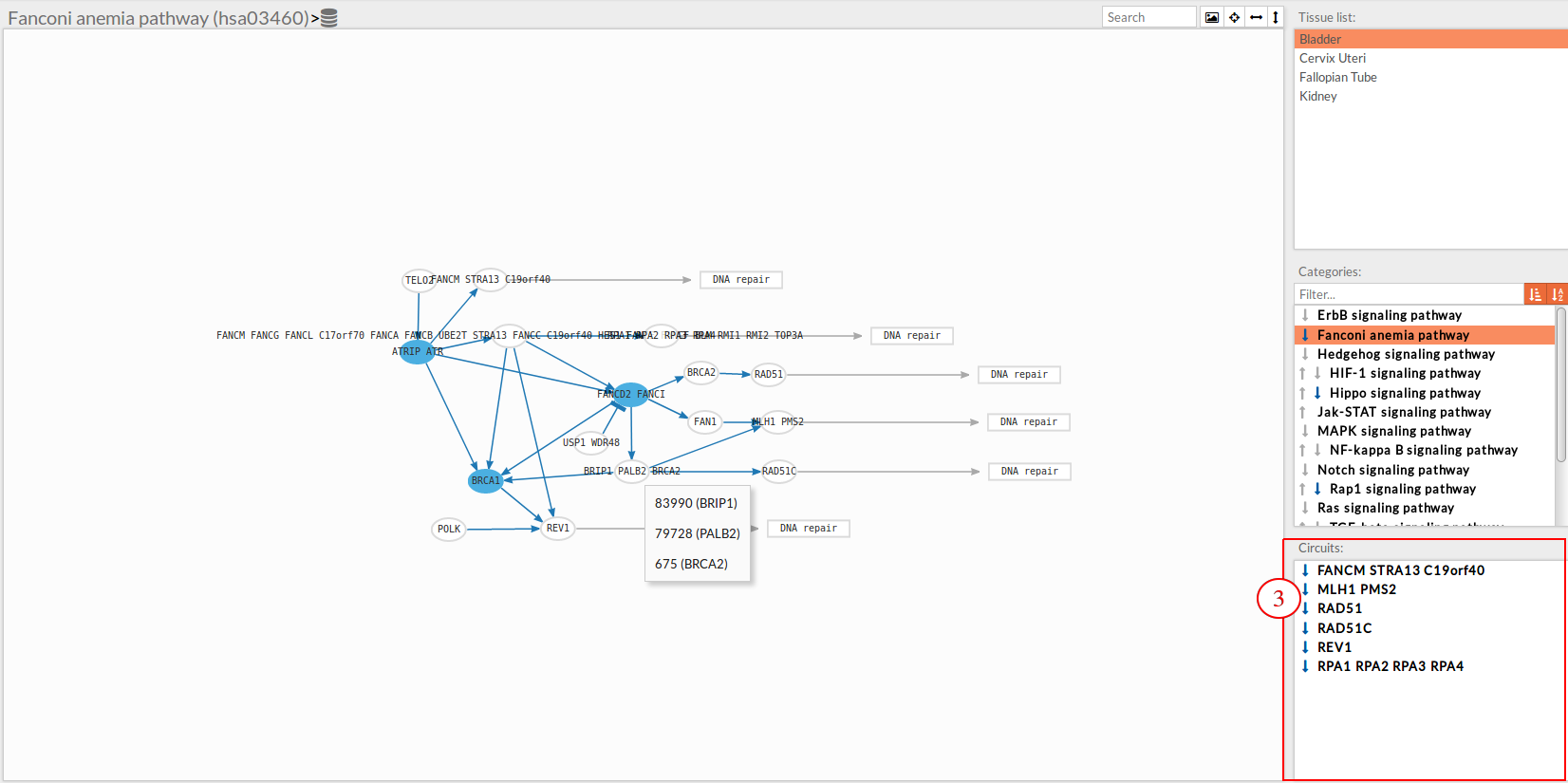
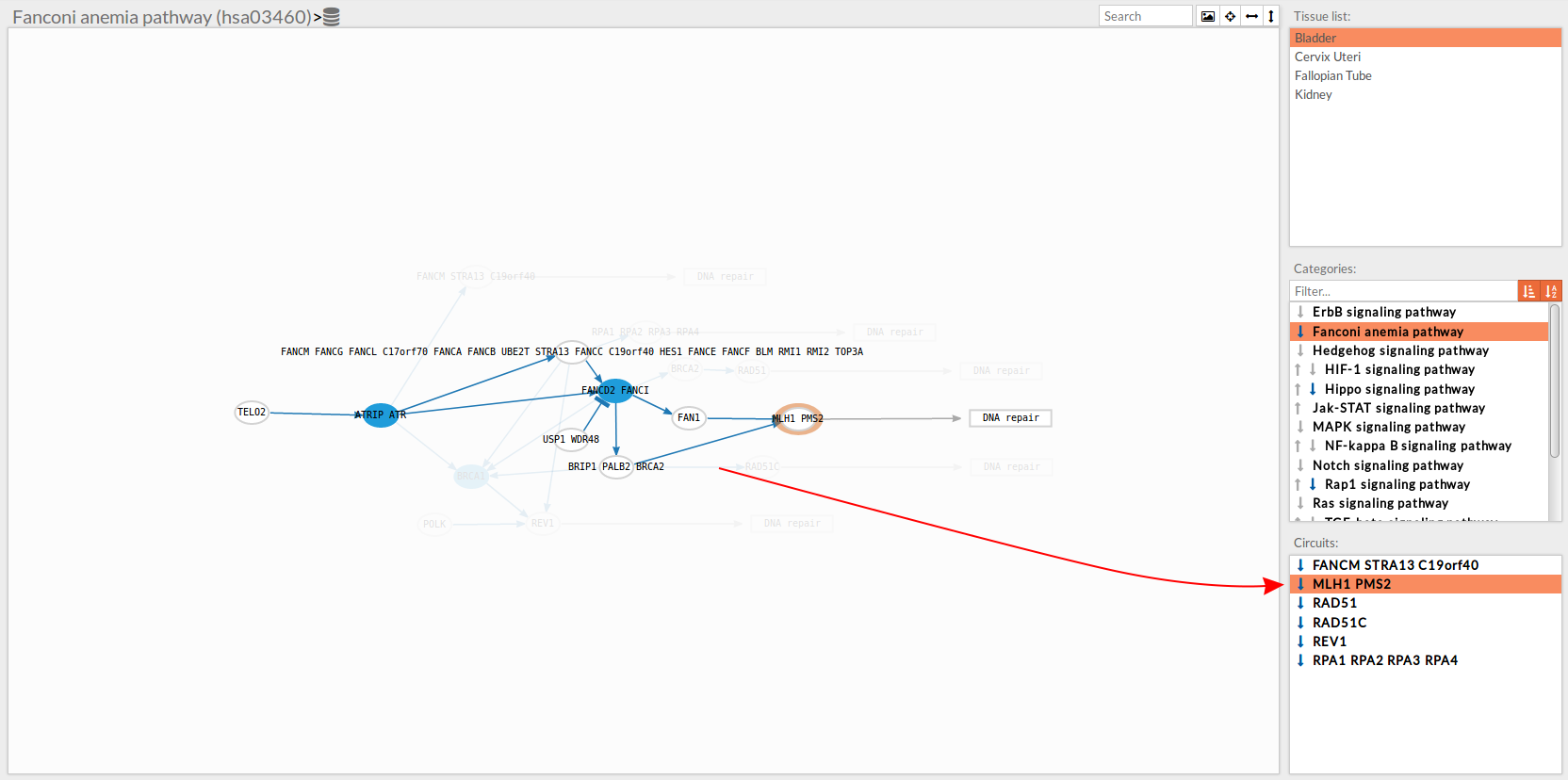
In the lower-right part of the tool will appear all the circuits in which a pathway (previously selected on the upper part -2-) can be decomposed.
 Once any of the circuit is clicked the nodes and interactions (edges) that form part of this circuit are highlighted in the pathway viewer, One example might be the blue-highlighted circuit on the figure below.
Once any of the circuit is clicked the nodes and interactions (edges) that form part of this circuit are highlighted in the pathway viewer, One example might be the blue-highlighted circuit on the figure below.
In the visualization part there are two types of objects, the nodes and the edges.
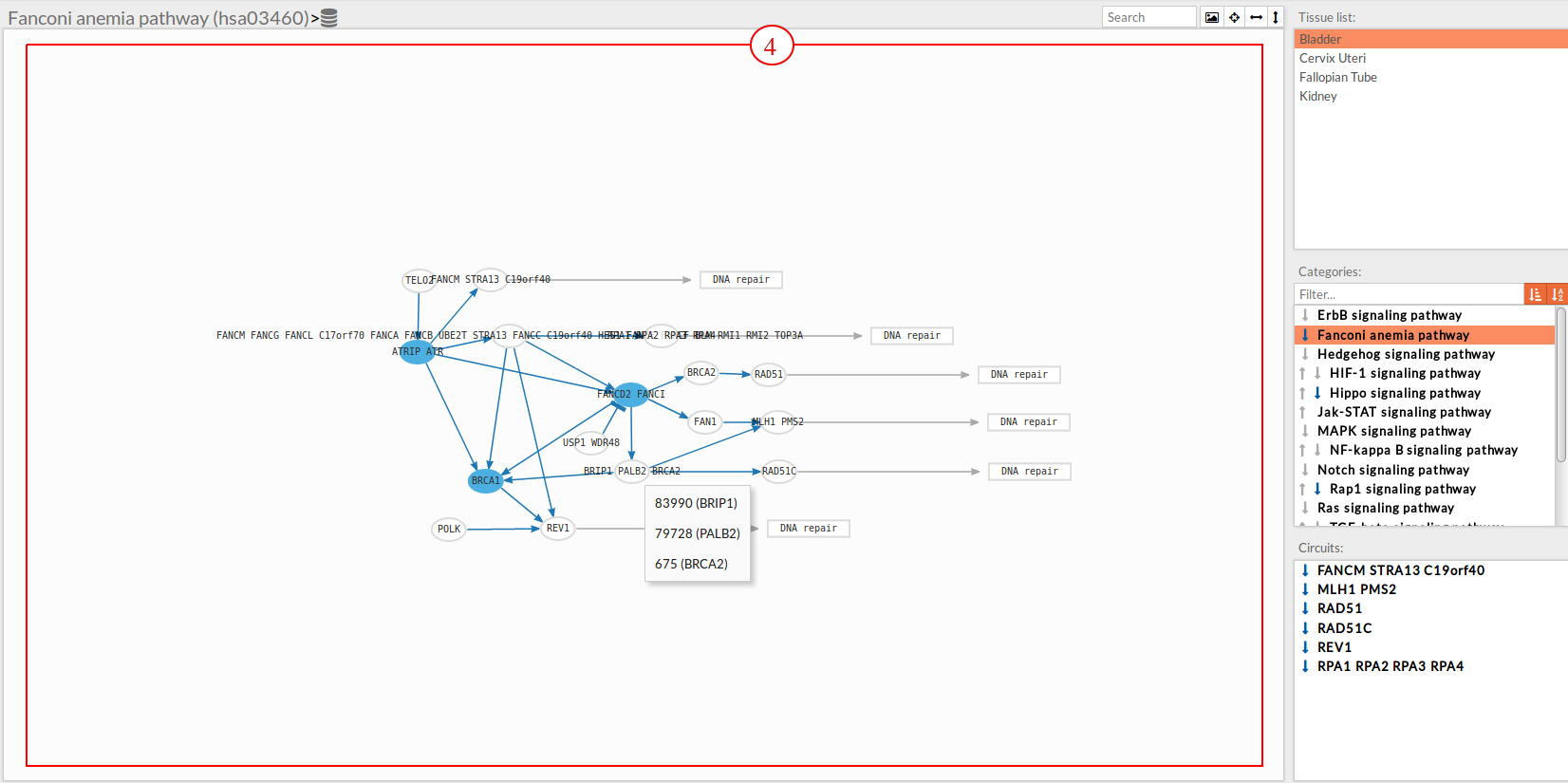
 The top part contains the title of selected pathway,by clicking on this button
The top part contains the title of selected pathway,by clicking on this button  you can see the original source of this pathway.
Also you can find other button in the right side:
you can see the original source of this pathway.
Also you can find other button in the right side:
 Allow to search specific genes, proteins or functions.
Allow to search specific genes, proteins or functions. Export a SVG image of viewed objects (the whole pathway or just the selected effector circuit)
Export a SVG image of viewed objects (the whole pathway or just the selected effector circuit) Center the selected pathway
Center the selected pathway Width adjust
Width adjust  Height adjust
Height adjust 